
Selected publications
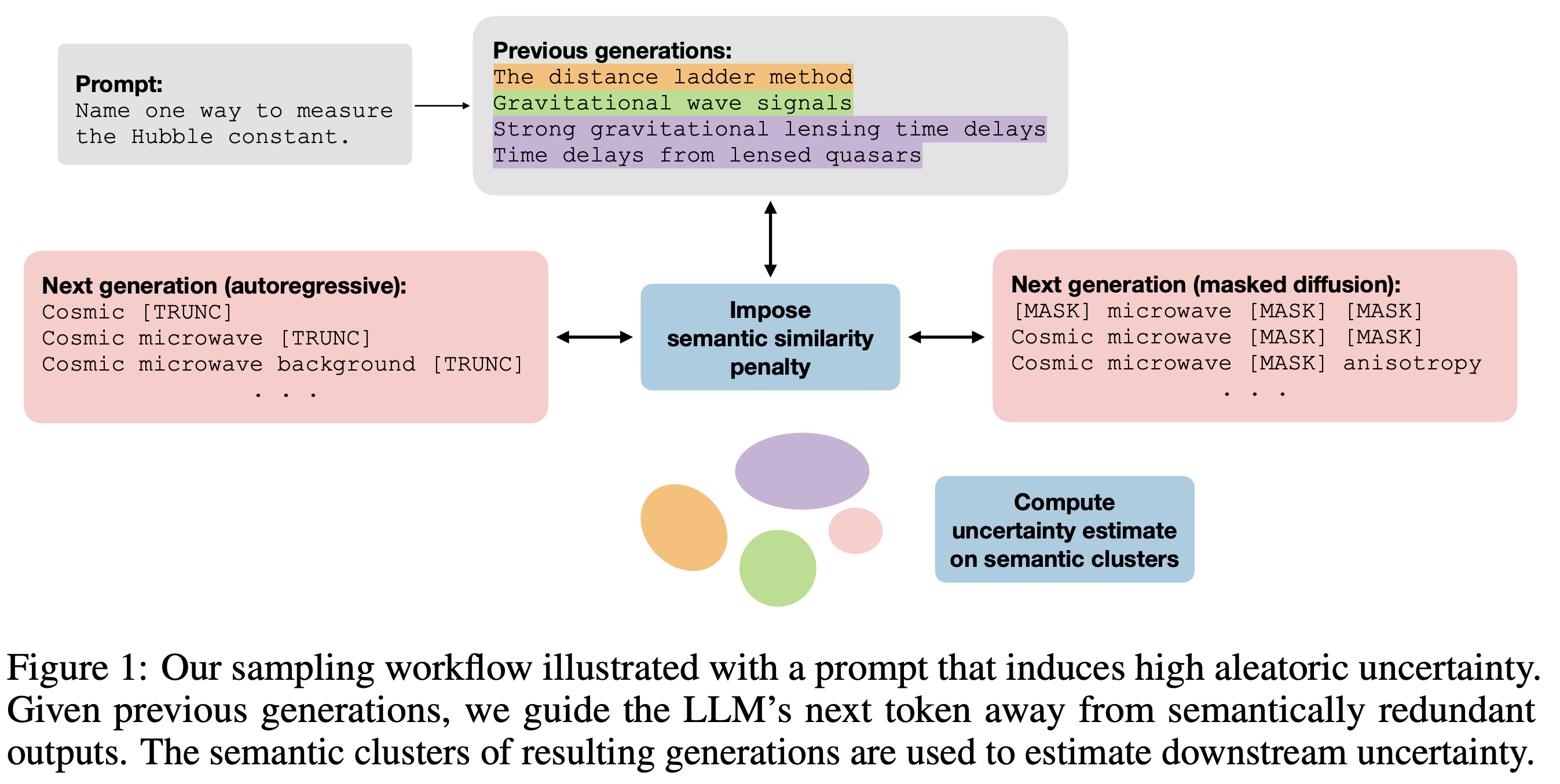
Efficient semantic uncertainty quantification in language models via diversity-steered sampling
NeurIPS (2025)[arXiv]
Abstract
Accurately estimating semantic aleatoric and epistemic uncertainties in large language models (LLMs) is particularly challenging in free-form question answering (QA), where obtaining stable estimates often requires many expensive generations. We introduce a diversity-steered sampler that discourages semantically redundant outputs during decoding, covers both autoregressive and masked diffusion paradigms, and yields substantial sample-efficiency gains. The key idea is to inject a continuous semantic-similarity penalty into the model's proposal distribution using a natural language inference (NLI) model lightly finetuned on partial prefixes or intermediate diffusion states. We debias downstream uncertainty estimates with importance reweighting and shrink their variance with control variates. Across four QA benchmarks, our method matches or surpasses baselines while covering more semantic clusters with the same number of samples. Being modular and requiring no gradient access to the base LLM, the framework promises to serve as a drop-in enhancement for uncertainty estimation in risk-sensitive model deployments.
BibTeX
@inproceedings{
park2025efficient,
title={Efficient semantic uncertainty quantification in language models via diversity-steered sampling},
author={Ji Won Park and Kyunghyun Cho},
booktitle={The Thirty-ninth Annual Conference on Neural Information Processing Systems},
year={2025},
url={https://openreview.net/forum?id=IiEtQPGVyV}
}
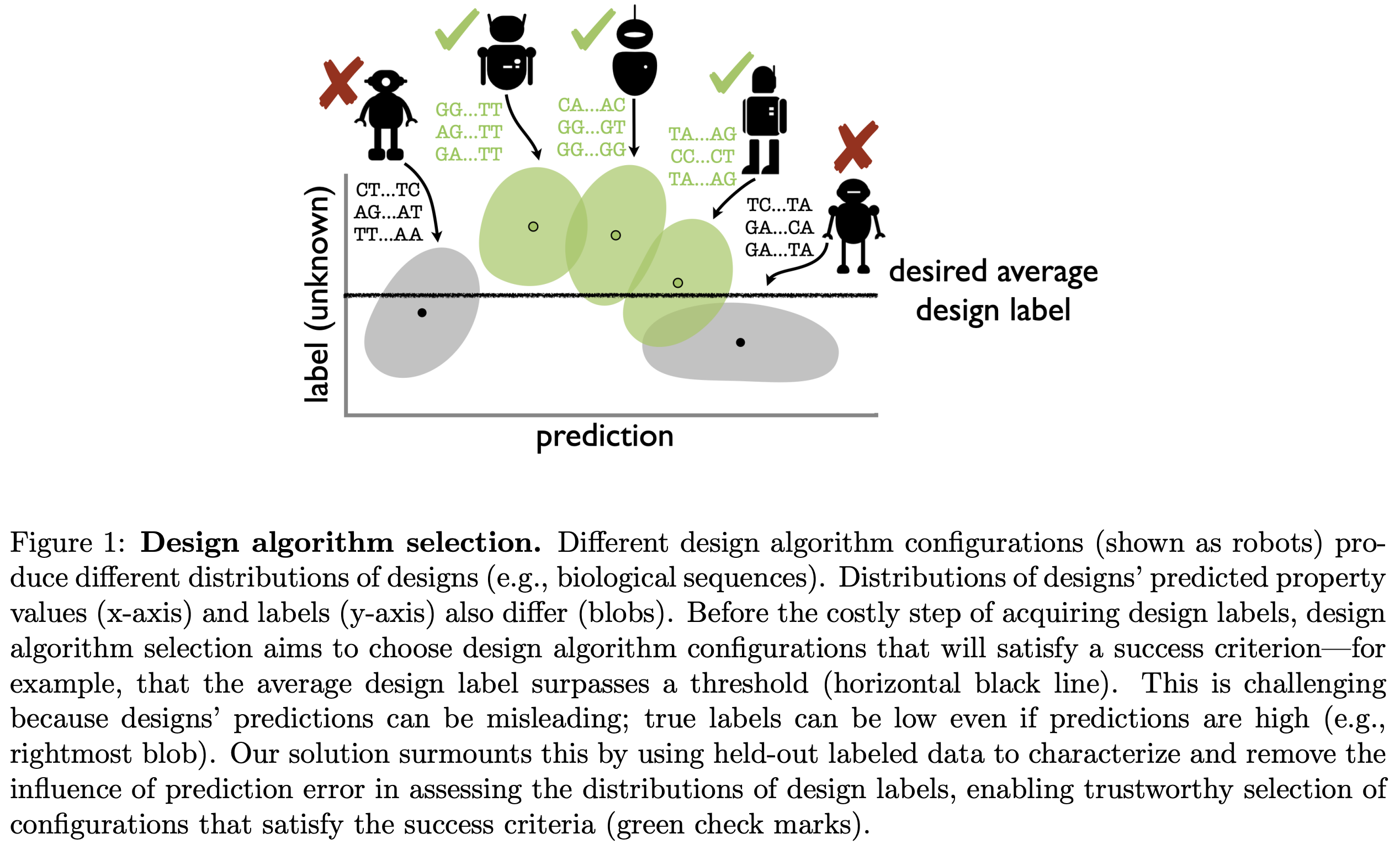
Reliable algorithm selection for machine learning-guided design
ICML (2025)[arXiv]
Abstract
Algorithms for machine learning-guided design, or design algorithms, use machine learning-based predictions to propose novel objects with desired property values. Given a new design task -- for example, to design novel proteins with high binding affinity to a therapeutic target -- one must choose a design algorithm and specify any hyperparameters and predictive and/or generative models involved. How can these decisions be made such that the resulting designs are successful? This paper proposes a method for design algorithm selection, which aims to select design algorithms that will produce a distribution of design labels satisfying a user-specified success criterion -- for example, that at least ten percent of designs' labels exceed a threshold. It does so by combining designs' predicted property values with held-out labeled data to reliably forecast characteristics of the label distributions produced by different design algorithms, building upon techniques from prediction-powered inference. The method is guaranteed with high probability to return design algorithms that yield successful label distributions (or the null set if none exist), if the density ratios between the design and labeled data distributions are known. We demonstrate the method's effectiveness in simulated protein and RNA design tasks, in settings with either known or estimated density ratios.
BibTeX
@inproceedings{
fannjiang2025reliable,
title={Reliable Algorithm Selection for Machine Learning-Guided Design},
author={Clara Fannjiang and Ji Won Park},
booktitle={Forty-second International Conference on Machine Learning},
year={2025},
url={https://openreview.net/forum?id=mFMGldHdOo}
}
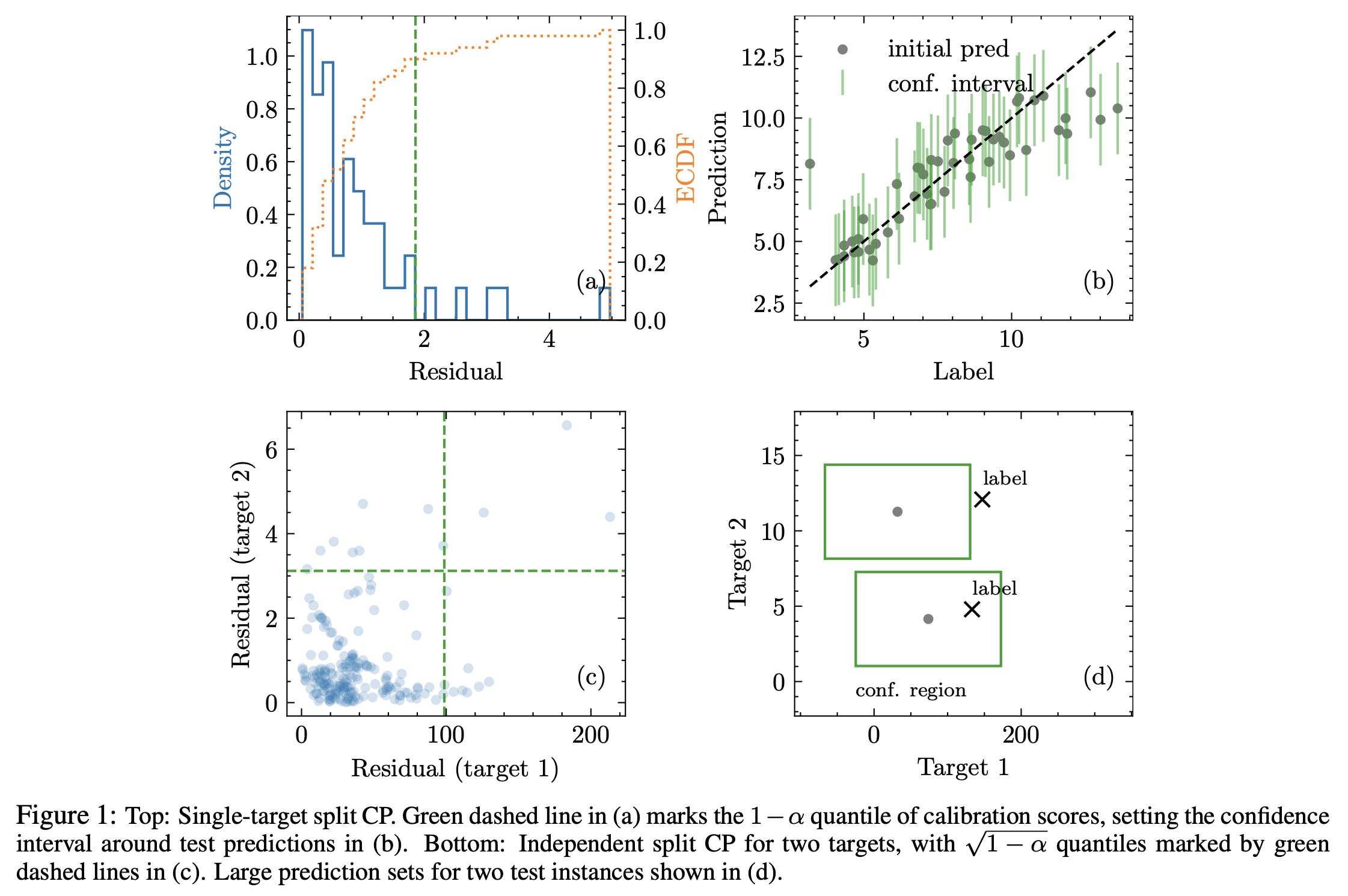
Semiparametric conformal prediction
AISTATS (2024)[arXiv]
Abstract
Many risk-sensitive applications require well-calibrated prediction sets over multiple, potentially correlated target variables, for which the prediction algorithm may report correlated errors. In this work, we aim to construct the conformal prediction set accounting for the joint correlation structure of the vector-valued non-conformity scores. Drawing from the rich literature on multivariate quantiles and semiparametric statistics, we propose an algorithm to estimate the 1-α quantile of the scores, where α is the user-specified miscoverage rate. In particular, we flexibly estimate the joint cumulative distribution function (CDF) of the scores using nonparametric vine copulas and improve the asymptotic efficiency of the quantile estimate using its influence function. The vine decomposition allows our method to scale well to a large number of targets. As well as guaranteeing asymptotically exact coverage, our method yields desired coverage and competitive efficiency on a range of real-world regression problems, including those with missing-at-random labels in the calibration set.
BibTeX
@inproceedings{
park2025semiparametric,
title={Semiparametric conformal prediction},
author={Ji Won Park and Kyunghyun Cho},
booktitle={The 28th International Conference on Artificial Intelligence and Statistics},
year={2025},
url={https://openreview.net/forum?id=ENscW7Y0lr}
}
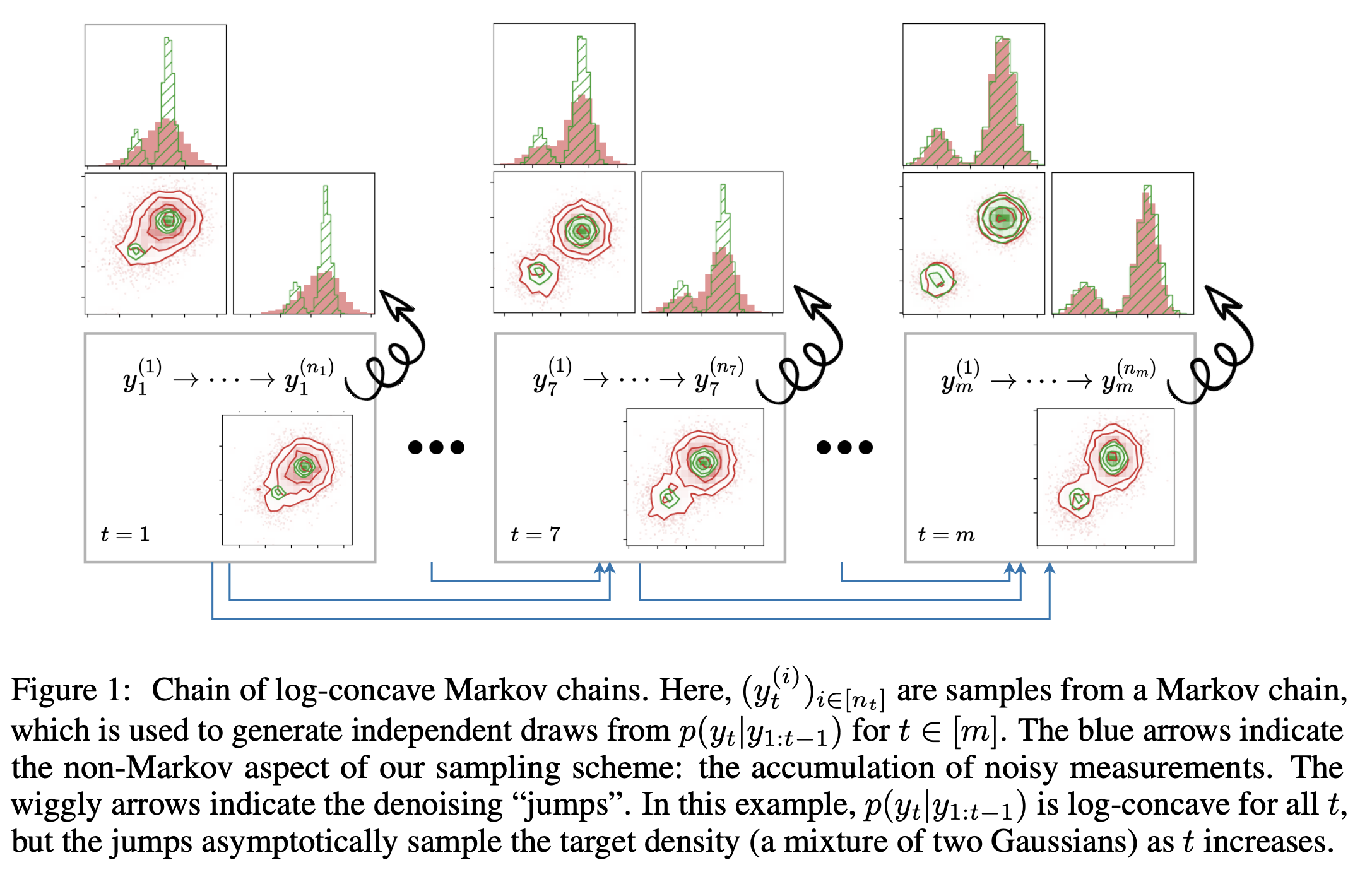
Chain of Log-Concave Markov Chains
ICLR (2024)[arXiv]
Abstract
We introduce a theoretical framework for sampling from unnormalized densities based on a smoothing scheme that uses an isotropic Gaussian kernel with a single fixed noise scale. We prove one can decompose sampling from a density (minimal assumptions made on the density) into a sequence of sampling from log-concave conditional densities via accumulation of noisy measurements with equal noise levels. Our construction is unique in that it keeps track of a history of samples, making it non-Markovian as a whole, but it is lightweight algorithmically as the history only shows up in the form of a running empirical mean of samples. Our sampling algorithm generalizes walk-jump sampling (Saremi & Hyvärinen, 2019). The "walk" phase becomes a (non-Markovian) chain of (log-concave) Markov chains. The "jump" from the accumulated measurements is obtained by empirical Bayes. We study our sampling algorithm quantitatively using the 2-Wasserstein metric and compare it with various Langevin MCMC algorithms. We also report a remarkable capacity of our algorithm to "tunnel" between modes of a distribution.
BibTeX
@inproceedings{
saremi2024chain,
title={Chain of Log-Concave Markov Chains},
author={Saeed Saremi and Ji Won Park and Francis Bach},
booktitle={The Twelfth International Conference on Learning Representations},
year={2024},
url={https://openreview.net/forum?id=yiMB2DOjsR}
}
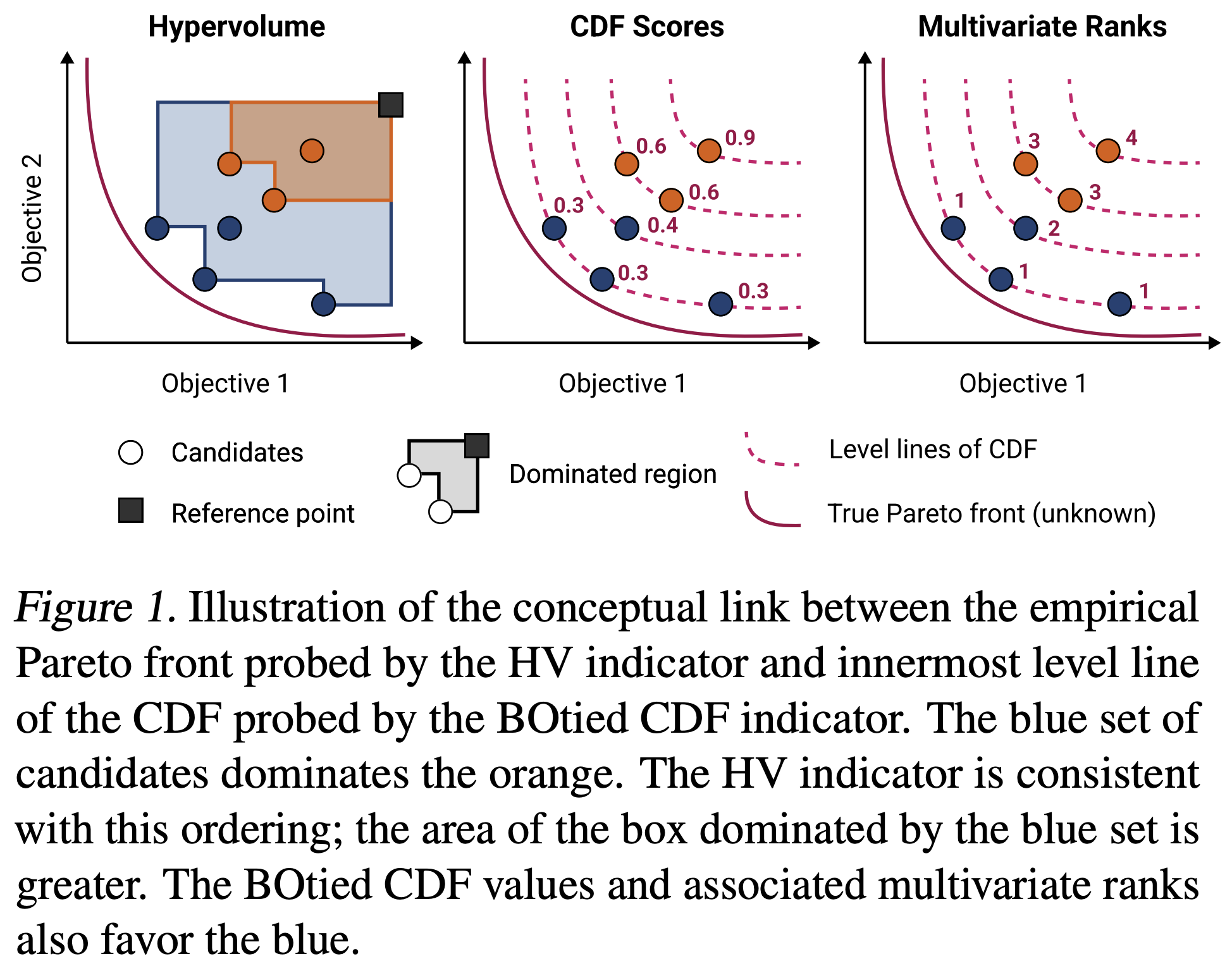
BOtied: Multi-objective Bayesian optimization with tied multivariate ranks
ICML (2024)[arXiv]
Abstract
Many scientific and industrial applications require the joint optimization of multiple, potentially competing objectives. Multi-objective Bayesian optimization (MOBO) is a sample-efficient framework for identifying Pareto-optimal solutions. At the heart of MOBO is the acquisition function, which determines the next candidate to evaluate by navigating the best compromises among the objectives. In this paper, we show a natural connection between non-dominated solutions and the extreme quantile of the joint cumulative distribution function (CDF). Motivated by this link, we propose the Pareto-compliant CDF indicator and the associated acquisition function, BOtied. BOtied inherits desirable invariance properties of the CDF, and an efficient implementation with copulas allows it to scale to many objectives. Our experiments on a variety of synthetic and real-world problems demonstrate that BOtied outperforms state-of-the-art MOBO acquisition functions while being computationally efficient for many objectives.
BibTeX
@inproceedings{
park2024botied,
title={{BO}tied: Multi-objective Bayesian optimization with tied multivariate ranks},
author={Ji Won Park and Natasa Tagasovska and Michael Maser and Stephen Ra and Kyunghyun Cho},
booktitle={Forty-first International Conference on Machine Learning},
year={2024},
url={https://openreview.net/forum?id=cj5HbaX14p}
}
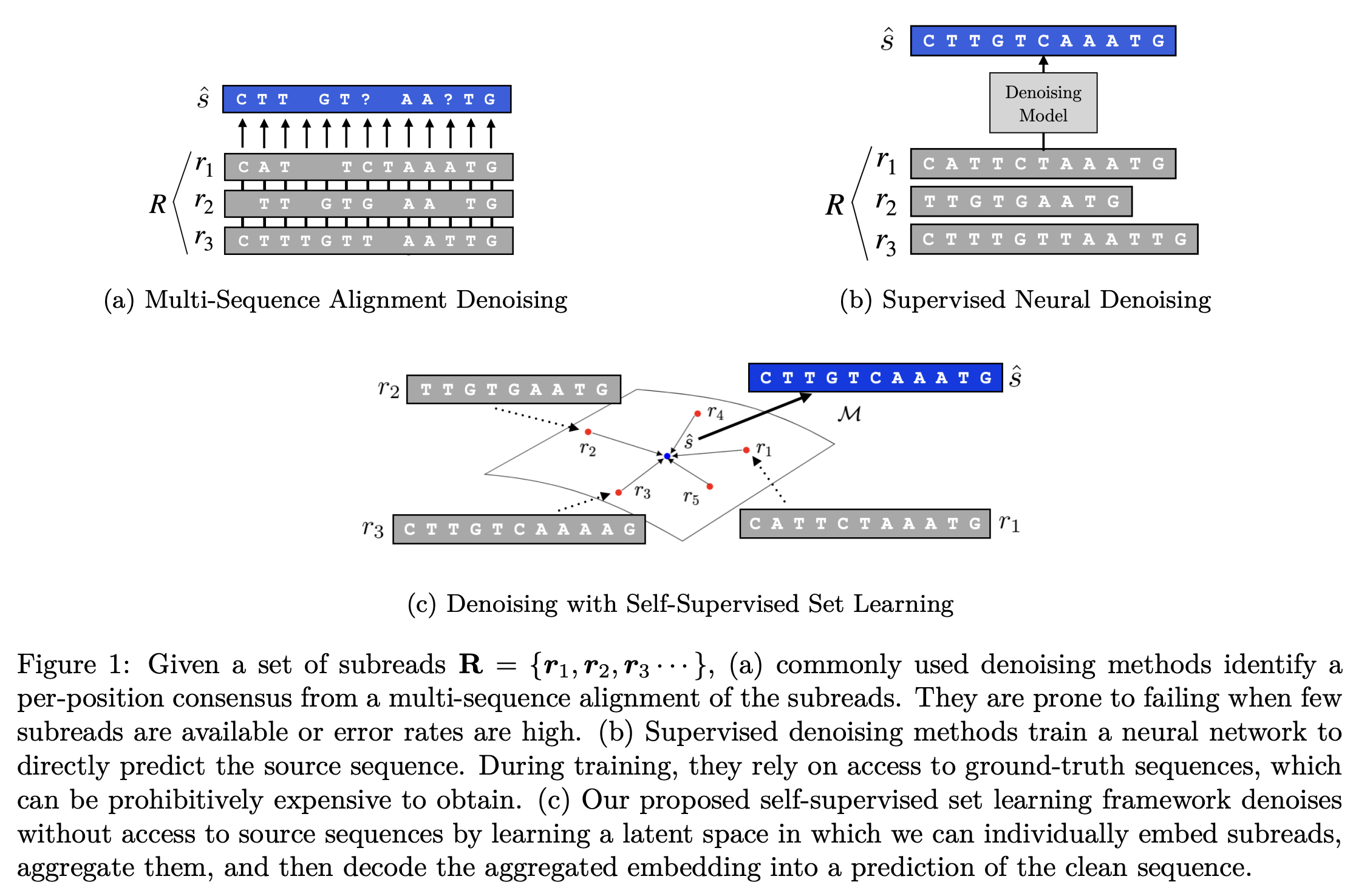
Blind Biological Sequence Denoising with Self-Supervised Set Learning
TMLR (2024)[arXiv]
Abstract
Biological sequence analysis relies on the ability to denoise the imprecise output of sequencing platforms. We consider a common setting where a short sequence is read out repeatedly using a high-throughput long-read platform to generate multiple subreads, or noisy observations of the same sequence. Denoising these subreads with alignment-based approaches often fails when too few subreads are available or error rates are too high. In this paper, we propose a novel method for blindly denoising sets of sequences without directly observing clean source sequence labels. Our method, Self-Supervised Set Learning (SSSL), gathers subreads together in an embedding space and estimates a single set embedding as the midpoint of the subreads in both the latent and sequence spaces. This set embedding represents the "average" of the subreads and can be decoded into a prediction of the clean sequence. In experiments on simulated long-read DNA data, SSSL methods denoise small reads of ≤ 6 subreads with 17% fewer errors and large reads of > 6 subreads with 8% fewer errors compared to the best baseline. On a real dataset of antibody sequences, SSSL improves over baselines on two self-supervised metrics, with a significant improvement on difficult small reads that comprise over 60% of the test set. By accurately denoising these reads, SSSL promises to better realize the potential of high-throughput DNA sequencing data for downstream scientific applications.
BibTeX
@article{
ng2024blind,
title={Blind Biological Sequence Denoising with Self-Supervised Set Learning},
author={Nathan Hoyen Ng and Ji Won Park and Jae Hyeon Lee and Ryan Lewis Kelly and Stephen Ra and Kyunghyun Cho},
journal={Transactions on Machine Learning Research},
issn={2835-8856},
year={2024},
url={https://openreview.net/forum?id=3s7ior0WZ5},
}
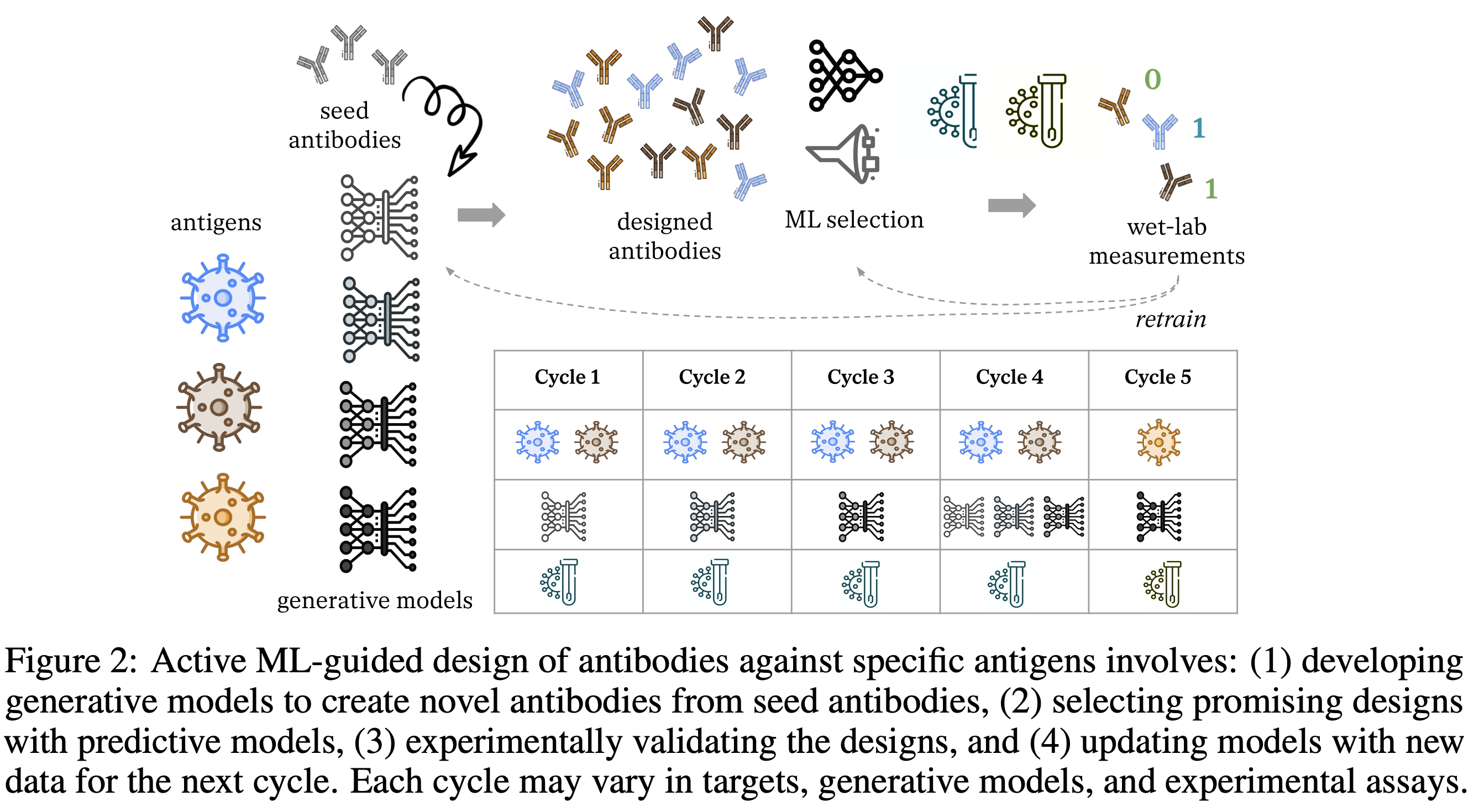
Antibody DomainBed: Out-of-Distribution Generalization in Therapeutic Protein Design
Oral Presentation, Workshop on Spurious Correlations, Invariance and Stability (SCIS) at ICML 2023 (2024)[arXiv]
Abstract
Machine learning (ML) has demonstrated significant promise in accelerating drug design. Active ML-guided optimization of therapeutic molecules typically relies on a surrogate model predicting the target property of interest. The model predictions are used to determine which designs to evaluate in the lab, and the model is updated on the new measurements to inform the next cycle of decisions. A key challenge is that the experimental feedback from each cycle inspires changes in the candidate proposal or experimental protocol for the next cycle, which lead to distribution shifts. To promote robustness to these shifts, we must account for them explicitly in the model training. We apply domain generalization (DG) methods to classify the stability of interactions between an antibody and antigen across five domains defined by design cycles. Our results suggest that foundational models and ensembling improve predictive performance on out-of-distribution domains. We publicly release our codebase extending the DG benchmark "DomainBed," and the associated dataset of antibody sequences and structures emulating distribution shifts across design cycles.
BibTeX
@article{tagasovska2024antibody,
title={Antibody DomainBed: Out-of-Distribution Generalization in Therapeutic Protein Design},
author={Tagasovska, Natasa and Park, Ji Won and Kirchmeyer, Matthieu and Frey, Nathan C. and Watkins, Andrew Martin and Ismail, Aya Abdelsalam and Jamasb, Arian Rokkum and Lee, Edith and Bryson, Tyler and Ra, Stephen and Cho, Kyunghyun},
journal={arXiv preprint arXiv:2407.21028},
year={2024}
}